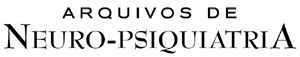
LETTER
Hypergonadotropic hypogonadism and cerebellar ataxia: an unusual association
Hipogonadismo hipergonadotrófico e ataxia cerebelar: uma associação rara
Pedro Braga-NetoI; Maria Cecília Resende MartinsII; Denizart Santos- NetoI; Patrícia WeismanI; Elaine de Paula Fiod CostaIII; Luciano Moreira PintoIII; Ruth Ferreira Santos-GaldurozIII; Ieda Therezinha do Nascimento VerreschiII; Orlando Graziani Povoas BarsottiniI
IFederal University of São Paulo (UNIFESP), São Paulo SP, Brazil: Department of Neurology and Neurosurgery
IIFederal University of São Paulo (UNIFESP), São Paulo SP, Brazil: Department of Endocrinology
IIIFederal University of São Paulo (UNIFESP), São Paulo SP, Brazil: Department of Ophthalmology
Correspondence Correspondence Pedro Braga Neto Rua Leandro Dupre 488 / 173 04025-012 São Paulo SP - Brasil E-mail: pbraganeto@hotmail.com
The association between cerebellar ataxia and hypogonadism was first described by Gordon Holmes in 19071. It represents a highly heterogenous syndrome with insidious onset. The hypogonadism of most patients with Gordon Holmes syndrome is hypogonadotropic, with a defect in the production or release of gonadotropins by pituitary glands2. In contrast, some patients with Holmes type ataxia may have hypergonadotropic hypogonadism, which represents a failure at the level of the gonads rather than at the level of the pituitary.
CASE
A 47-year-old woman presented to a neurology department with a nine- year history of progressive worsening balance. She had been a normal full term delivery of nonconsanguineos parents and presented delayed psychomotor and pubertal development, as well as primary amenorrhea. Tanner stage was 4/5, pubic hair 5/5 and axillary hair 3/3. Her height and weight was normal.
On neurological examination, the muscle strength was 5/5 with slightly brisk tendon reflexes. There was an ataxic gait with mildly broad based lower limbs, considerable staggering and difficult with half turn. She was unable to walk tandem (heel-to-toe). Finger-nose and heel-shin ataxia were evident with clumsiness of fine finger movements. There was moderate overshoot on upper limb ballistic tracking movements. Gaze-evoked horizontal nystagmus, cleary ocular saccadic overshot and cerebellar dysartria were also present. The symptoms slightly progressed throughout the years. Fundoscopy showed pigmentary retinopathy (Fig 1).
To score the cerebellar dysfunction, we used the International Cooperative Ataxia Rating Scale (ICARS) 3. The patient obtained a 49 score.
The patient was also submitted to the neuropsychological assessment composed of evaluation of intellectual level, visuospacial constructions, memory systems, executive functions, sustained attention, abstraction, cognitive flexibility, response inhibition, selective attention and concentration, language and capacity of abstraction. In relation to the expected scores of cognitive evaluation, the patient presented damage below the expected in the long term memory (declarative: episodic and semantic - this last one could be result, in part, of low stimulation), short term memory (and operational memory), even so presents information capacity, it is located in elementary level. Also presented damage in fine motor skills (related to the agility), in visual- spacial ability, beyond the reduced mental flexibility and the abstraction capacity. In the impulse control test, the evaluation presented damage in the performance
Brain magnetic resonance imaging revealed a marked cerebellar atrophy and cerebellar peduncle atrophy (Fig 2). Karyotype was 46,XX and specific serum biochemistry was as follows: CPK: 76 U/L (26-155 U/L); vitamin E: 0.5 mg/dL (0.5-1.8 mg/dL); lactic acid: 11.82 mg/dL (<22 mg/dL); aldolase 3.8 mg/dL (<7.4 mg/dL); piruvic acid 0.04 mmol/L (0.03-0.1mmol/L). Routine blood and urine testing, echocardiogram and electrocardiogram were all normal. The audiogram showed normal hearing.
Endocrinological studies were as follows: TSH 2.20 Uiu/ml (0.50-5.50 IU/ml); FSH 111.3 mIU/mL (post-menopausal range 23-116.3 mIU/mL); LH 39.30 mIU/mL (post-menopausal range 15.9-54 mIU/mL); estradiol <10.0 pg/mL(post-menopausal range <44.5 pg/mL); total testosterone 123 ng/dL (14-76 ng/dL); prolactin 11.1 ng/mL (post-menopausal rage 1.8-20.3 ng/mL).
An informed consent was obtained from the patient to allow data and images publication.
DISCUSSION
This case illustrates a condition characterized by adult onset ataxia, primary amenorrhea, hypergonadotropic hypogonadism, psychomotor retardation and pigmentary retinopathy.
Gordon Holmes was the first to describe the association between cerebellar ataxia and hypogonadism. Both hyper and hypogonadotropic forms were reported1. The disease has been designated as Gordon Holmes syndrome. However, the classification of this syndrome is very confusing and heterogenous. Some cases are designated as variants of Holmes type ataxia and others reported as a different entity. There is also little understanding of the pathophysiology mechanism of this association.
Seminara et al. reported the largest kindred and suggested that the disease is inherited as an autossomal recessive trait. The clinical and neurological evaluation of this group disclosed hypogonadotropic hypogonadism, progressive ataxia and dementia2.
Boucher and Gibberd in 1969 and Neuhausser and Opitz in 1975 described the association of spinocerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy as an autonomous single-gene disorder, designated as Boucher-Neuhauser syndrome4,5.
Amor et al. reported two sisters with adult onset cerebellar ataxia, hypergonadotropic hypogonadism, sensorineural deafness with vestibular hypofunction and normal intellect. He also suggested using a separate category of ataxia with hypergonadotropic hypogonadism6.
Primary coenzyme Q 10 deficiency is a rare and clinically heterogenous autossomal recessive caused by mutation in specific mitochondrial genes. The disorder has several major phenotypes, one of them a predominantly cerebellar form with ataxia and cerebellar atrophy7. Biochemically, the hallmark of the disease is a decreased coemzime Q 10 concentration in muscle and/or fibroblasts. The patient appeared to improve some cerebellar symptoms and perhaps, more markedly, some of the associated conditions8.
The association between hypogonadism hypogonadism, ataxia and sensoriomotor axonal polyneuropathy has also been described in a family of two siblings9.
A review of all reported cases of hypogonadism associated with cerebellar ataxia reveals a great heterogeneity in the causative pathogenic mechanisms. It is more reasonable to ascribe this association as a clinical entity with different genotypes involved, which may lead to the same or similar phenotypic manifestations.
Received 7 April 2009
Received in final form 15 July 2009
Accepted 11 September 2009
- 1. Holmes G. A form of familial degeneration of the cerebellum. Brain 1907; 30:466-489.
- 2. Seminara SB, Acierno Jr JS, Adulwahid NA, Crowley WFJr, Margolin DH. Hypogonadotropic hypogonadism and cerebellar ataxia: detailed phenotypic characterization of a large, extended kindred. J Clin Endocr Metab 2002;87:1607-1612.
- 3. Troillas P, Takayanagi T, Hallett M, et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. J Neurol Sci 1997;145:205-211.
- 4. Boucher BJ, Gibberd FB. Familial ataxia, hypogonadism and retinal degeneration. Acta Neurol Scand 1969;45:507-510.
- 5. Neuhauser G, Opitz JM. Autosomal recessive syndrome of cerebellar ataxia and hypogonadotropic hypogonadism. Clin Genet 1975;7:426-434.
- 6. Amor DJ, Delatycki MB, Gardner RJM, Storey E. New variant of familial cerebellar ataxia with hypergonadotrophic hypogonadism and sensorial deafness. Am J Med Genetics 2001;99:29-33.
- 7. Lamperti C, Naini A, Hirano M, et al. Cerebellar ataxia and coenzime Q10 deficiency. Neurology 2003;60:1206-1208.
- 8. Gironi M, Lamperti C, Nemni R, et al. Late-onser cerebellar ataxia with hypogonadism and muscle Q10 deficiency. Neurology 2004;62:818-820.
- 9. Erdemoglu AK, Akbostanci MC, Selçuki D. Familial cerebellar ataxia and hypogonadism associated with sensoriomotor axonal polyneuropathy. Clin Neurol Neurosurg 2000;102:129-134.
Correspondence
Publication Dates
-
Publication in this collection
22 Sept 2010 -
Date of issue
Feb 2010